
YANG CUI, Ph.D
About
I obtained my Ph.D. in Bioinformatics from the University of Tokyo, Department of Computational Biology and Medical Sciences, under the supervision of Professor Kenta Nakai. My research interests lie in cancer biology and computational biology, with a particular focus on the tumor microenvironment and spatial omics. During my Ph.D., I focused on dissecting the tumor microenvironment of triple-negative breast cancer (TNBC) through integrative multi-omics analysis. I am also actively involved in the development of computational tools for spatial transcriptomics analysis. My current work centers on building a computational framework to identify precancerous regions from spatial omics data.
Research Interests
Tumor Microenvironment and Spatial Omics

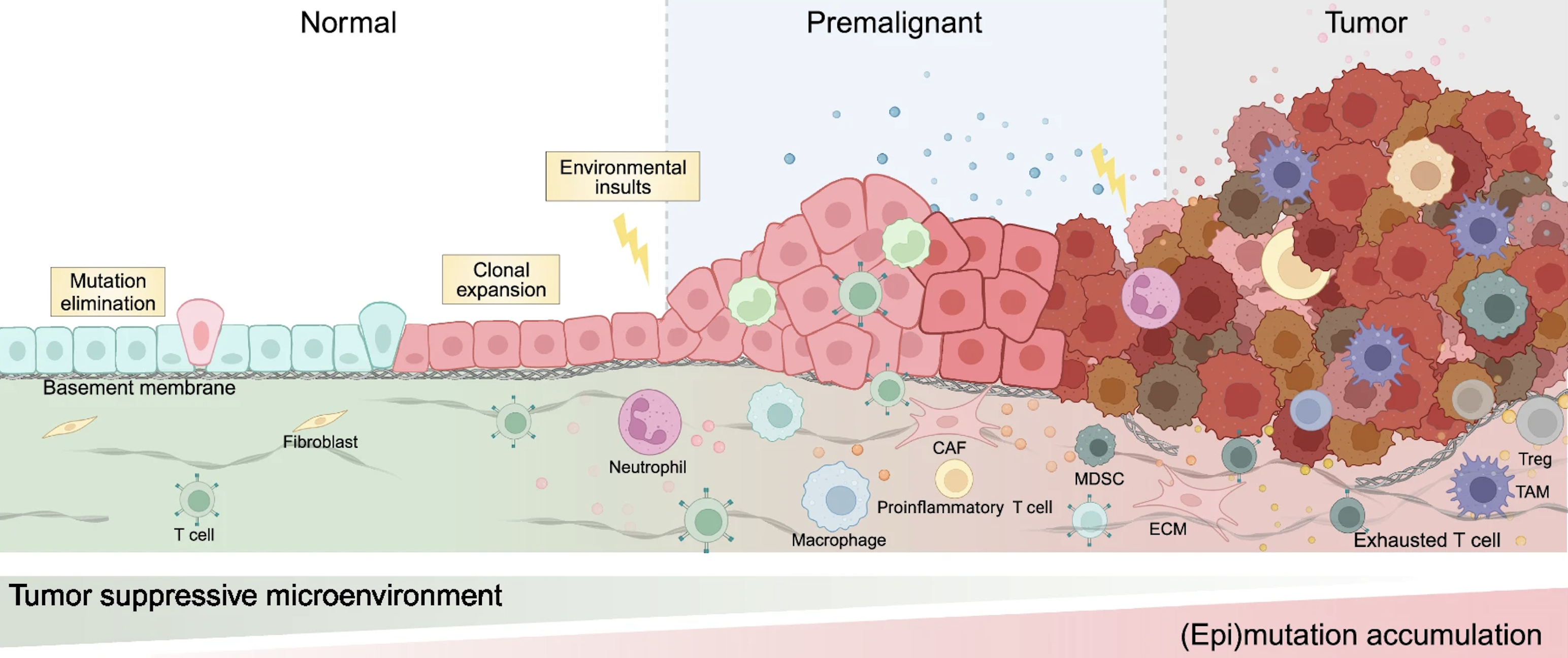
Our understanding of cancer has evolved beyond viewing it as a disease driven solely by malignant cells. Tumors operate within a complex tumor microenvironment (TME), consisting of immune cells, stromal cells and extracellular matrix components, which collectively influence tumor progression, metastasis, and immune evasion. Tumor cells can reshape the surrounding environment to promote their survival–for example, by inducing epithelial-mesenchymal transition (EMT), remodeling the extracellular matrix to enhance invasiveness and metastatic potential. They can also suppress key immune signals such as chemokines (e.g., CXCL9) and interleukins (e.g., IL-2), thereby inhibiting immune cell recruitment and activation. Even when immune cells are recruited, tumor cells can inhibit their function by expressing immune checkpoint molecules like PD-L1. The TME also contributes to the conversion of CD4⁺ T cells into regulatory T cells (Tregs), polarizes macrophages towards an M2-like phenotype, and drives CD8⁺ T cell exhaustion. Meanwhile, stromal cells such as fibroblasts within the TME can differentiate into cancer-associated fibroblasts (CAFs), which further support tumor growth and immune evasion.
To fully understand the TME, it is essential to elucidate the molecular profile of cancer. Large-scale bulk profiling projects, such as The Cancer Genome Atlas (TCGA), have established the foundation for understanding the molecular landscape of cancer. The emergence of single-cell omics has further advanced our understanding by revealing the cellular heterogeneity within tumors. Moreover, it has enabled the development of deconvolution methods (e.g., CIBERSORT) that retrospectively infer cell-type information from bulk datasets like TCGA, enhancing their biological interpretability. More recently, spatial omics has introduced a crucial missing dimension: spatial context. This allows us to investigate how tumor cells interact with their neighbors, how immune cells are spatially organized, and how niches within tumors drive differential responses to therapy.
My current research focuses on leveraging spatial omics technologies–including genomics, transcriptomics, epigenomics, and proteomics–to dissect the TME at unprecedented resolution. I aim to understand how tumor cells evolve within the TME, how they influence and are influenced by surrounding cells, and how these interactions shape therapeutic outcomes. Understanding these spatial interactions is particularly important in the context of immunotherapy, where patient responses remain highly variable. Given that only ~15% of patients benefit from PD-L1 blockade therapy, unraveling the spatial features of the TME may hold the key to identifying predictive biomarkers and guiding next-generation treatment strategies.
Detection of Spatially Resolved Precancerous Regions
(Zhang et al., STTT, 2024)
Deciphering how cancer evolves from normal tissue is fundamental to both basic cancer biology and clinical intervention. Environmental and genetic pressures, such as chronic inflammation, radiation exposure, or dysregulation of tumor suppressor genes like TP53 and BRCA1, can drive cells toward malignant transformation through a gradual accumulation of mutations. This process is often preceded by transcriptional and microenvironmental changes that occur before any histological abnormalities become visible.
The emergence of high-resolution spatial transcriptomics has made it possible to investigate cancer progression at cellular resolution within individual tumor sections. While previous studies have identified precancerous regions or transformation zones in cancer tissues, these discoveries often rely on manual annotation and prior biological knowledge, and currently there is no generalizable computational framework for discovering such regions in a data-driven way.
My current research focuses on developing computational methods to detect precancerous regions that are transcriptionally altered but morphologically indistinct. This work aims to uncover early spatial changes in tumorigenesis, improve our understanding of cancer heterogeneity, and ultimately contribute to earlier and more precise cancer diagnostics.